
Publications
Scientific publications from the Open Force Field Initiative.
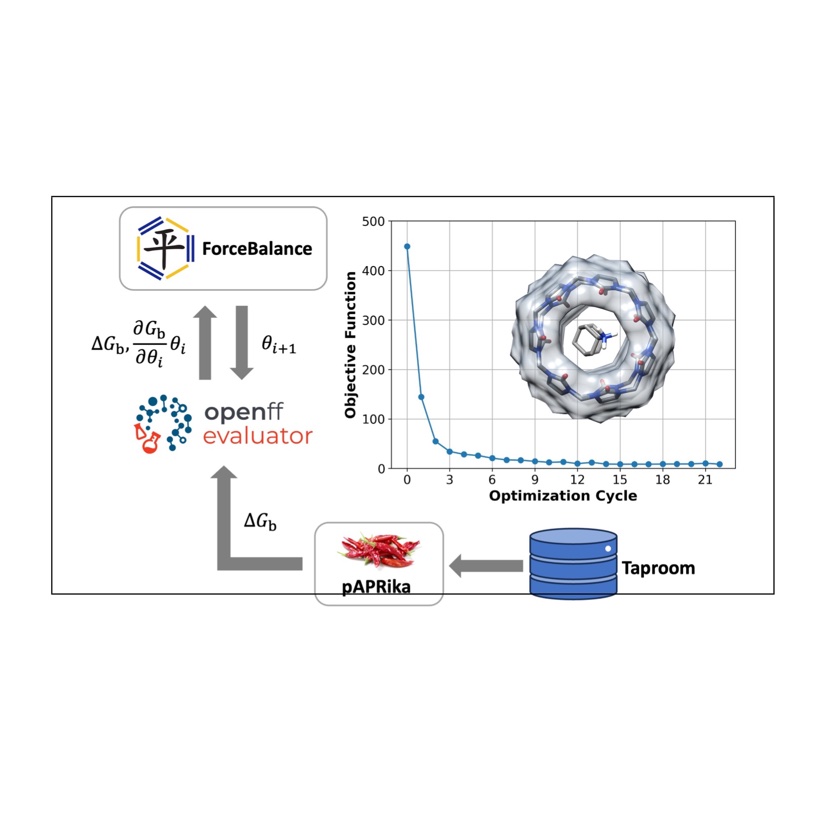
Tuning Potential Functions to Host–Guest Binding Data
Jeffry Setiadi, Simon Boothroyd, David Slochower, David Dotson, Matthew Thompson, Jeffrey Wagner, Lee-Ping Wang, and Michael K. Gilson
Preprint ahead of publication: chemRxiv
We extended the Open Force Field Evaluator framework to systematically calculate host-guest binding free energies, optimized force field parameters using experimental data, and demonstrated their potential transferability to protein-ligand systems.
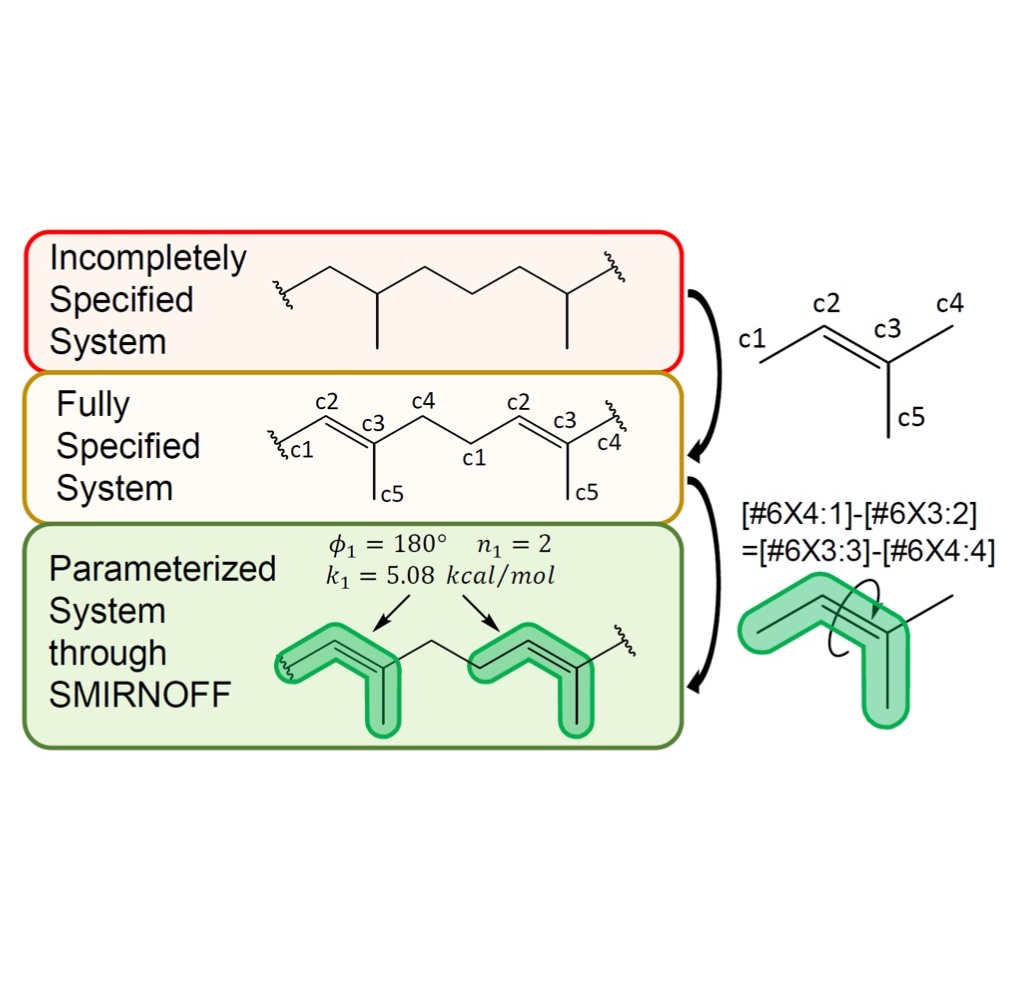
Parameterization of general organic polymers within the Open Force Field framework
Connor M. Davel, Timotej Bernat, Jeffrey R. Wagner, and Michael R. Shirts
Preprint ahead of publication: chemRxiv
We apply the Open Force Field framework to parameterize general organic polymers, demonstrating the breadth of chemical species that the SMIRNOFF approach can parameterize.
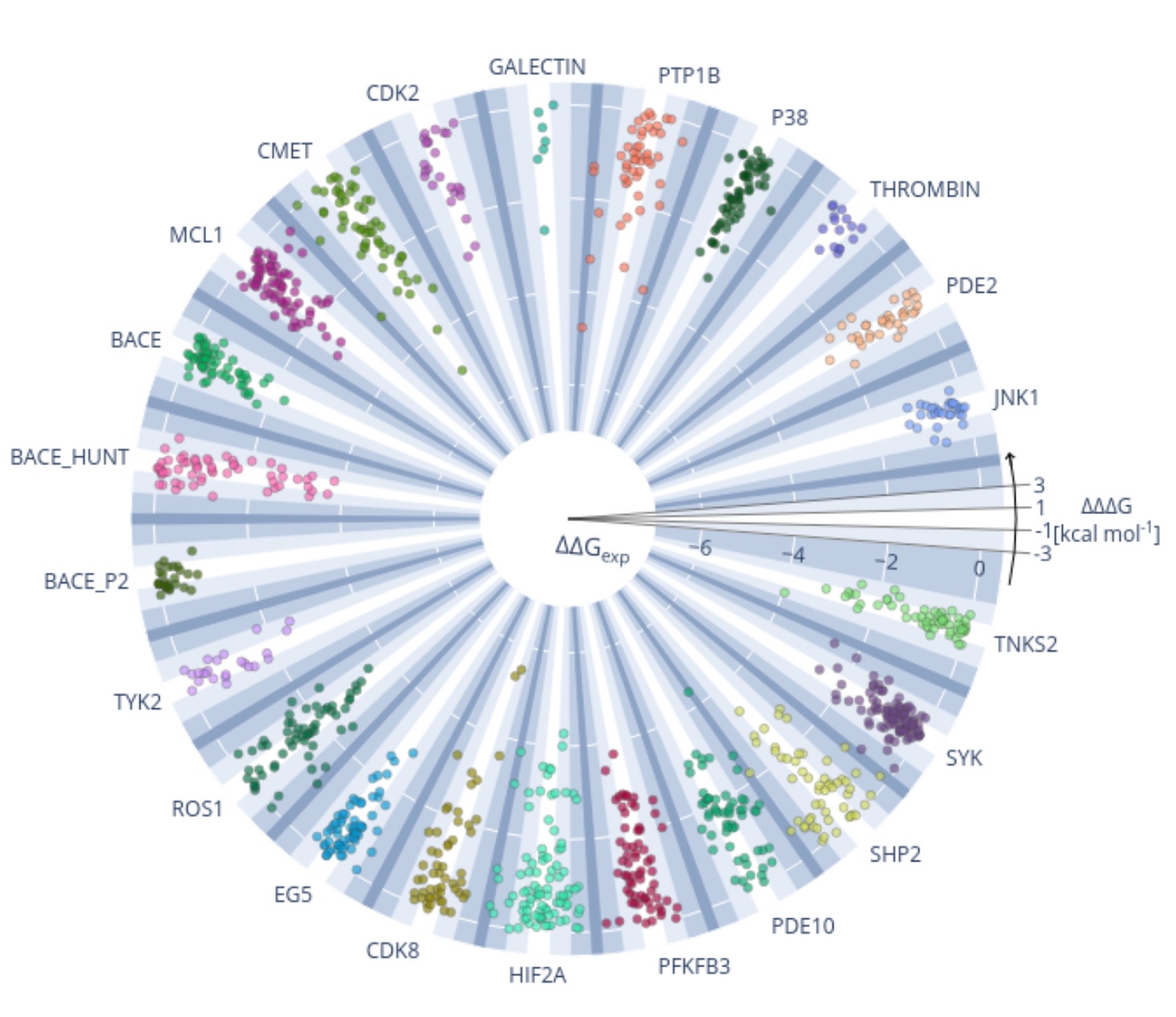
Current state of open source force fields in protein-ligand binding affinity predictions
David F. Hahn, Vytautas Gapsys, Bert L. de Groot, David L. Mobley, and Gary J. Tresadern
Preprint ahead of publication: chemRxiv
We benchmark the accuracy of OpenFF and major force fields for computing relative binding free energies on a set of 598 ligands and 22 protein targets.
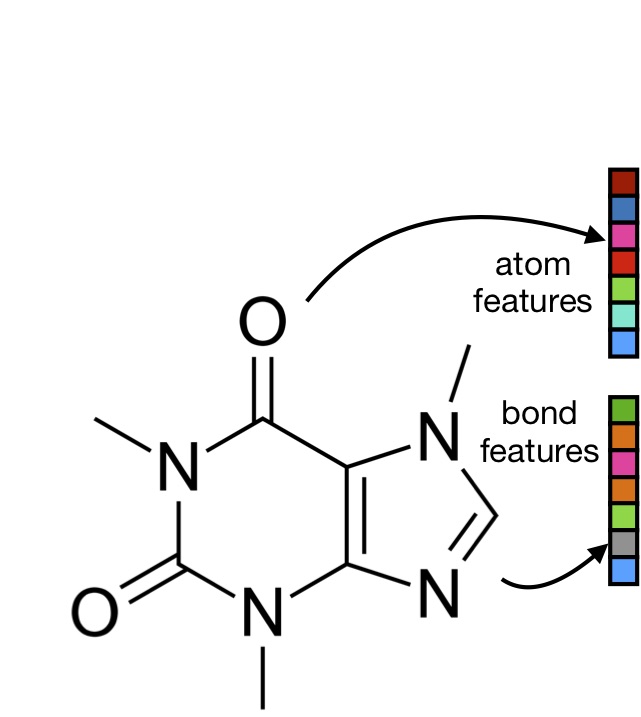
Machine-learned molecular mechanics force field for the simulation of protein-ligand systems and beyond
Kenichiro Takaba, Iván Pulido, Pavan Kumar Behara, Mike Henry, Hugo MacDermott-Opeskin, John D. Chodera, and Yuanqing Wang
Preprint ahead of publication: arXiv
Code accompanying the publication: choderalab/refit-espaloma
We show how the espaloma graph neural network can be used to construct a high-accuracy force field for protein-ligand systems from over 1.1 million quantum chemicl calculations, and demonstrate its performance on protein:ligand binding free energy calculations.
Hierarchical clustering of chemical space using binary-encoded SMARTS for building data-driven chemical perception models
Trevor Gokey and David L. Mobley
Preprint ahead of publication: chemRxiv
We demonstrate a method for automatically refining SMARTS-based typing for direct chemical perception to improve the accuracy of force fields.
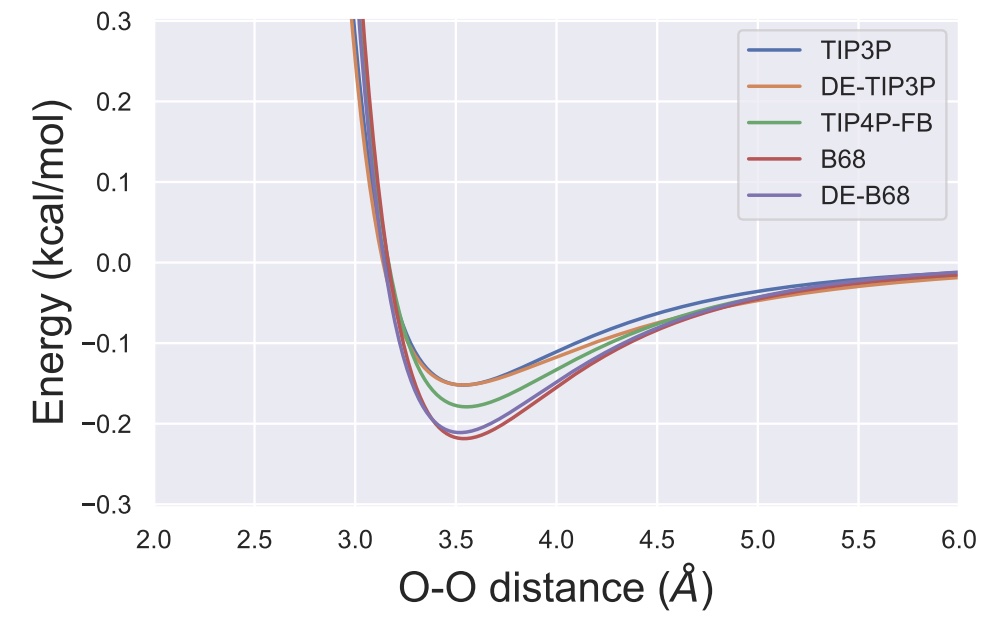
A transferable double exponential potential for condensed phase simulations of small molecules
Joshua T. Horton, Simon Boothroyd, Pavan Kumar Behara, David L. Mobley, and Daniel J. Cole
Preprint ahead of publication: chemRxiv
Published: Digital Discovery 2:1178, 2023 [DOI]
Code accompanying the publication: jthorton/de-forcefields
We show how the OpenFF Toolkit plugin scheme can be used to compare Lennard-Jones 12-6 with doubnle exponential potentials.
Structure-Based Experimental Datasets for Benchmarking of Protein Simulation Force Fields
Chapin E. Cavender, David A. Case, Julian C.-H. Chen, Lillian T. Chong, Daniel A. Keedy, Kresten Lindorff-Larsen, David L. Mobley, O. H. Samuli Ollila, Chris Oostenbrink, Paul Robustelli, Vincent A. Voelz, Michael E. Wall, David C. Wych, and Michael K. Gilson
Preprint ahead of publication: arXiv
We capture community consensus on best practices for assessing the quality of protein force fields via comparison with experimental data.
EspalomaCharge: Machine learning-enabled ultra-fast partial charge assignment
Yuanqing Wang, Iván Pulido, Kenichiro Takaba, Benjamin Kaminow, Jenke Scheen, Lily Wang, and John D. Chodera
Preprint ahead of publication: arXiv
Code accompanying the publication: choderalab/espaloma_charge
We present an ultra-fast, scalable graph net based model to generate AM1-BCC ELF10 quality charges self-consistently for small molecules and arbitrary biopolymers.
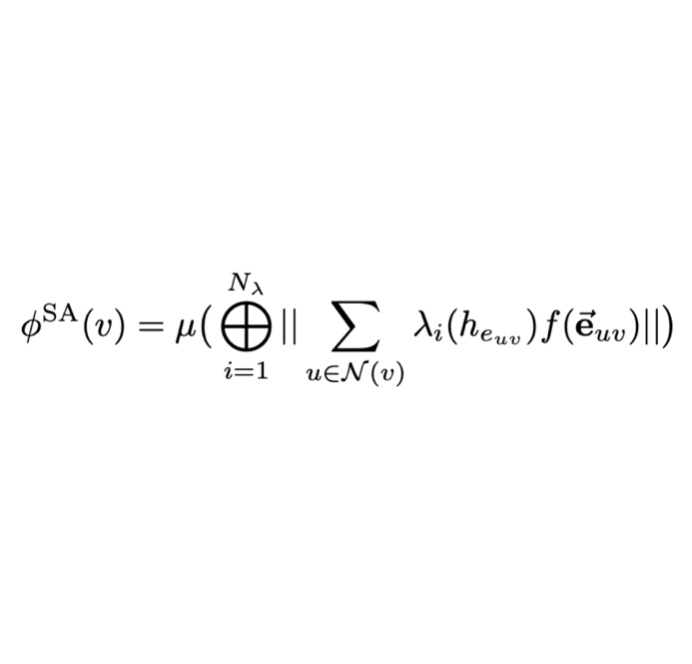
Spatial Attention Kinetic Networks with E(n)-Equivariance
Yuanqing Wang and John D. Chodera
Preprint ahead of publication: arXiv
Code accompanying the publication: choderalab/sake
We describe a new machine learning potential that provides an excellent tradeoff between speed and accuracy for learning quantum chemical energies.
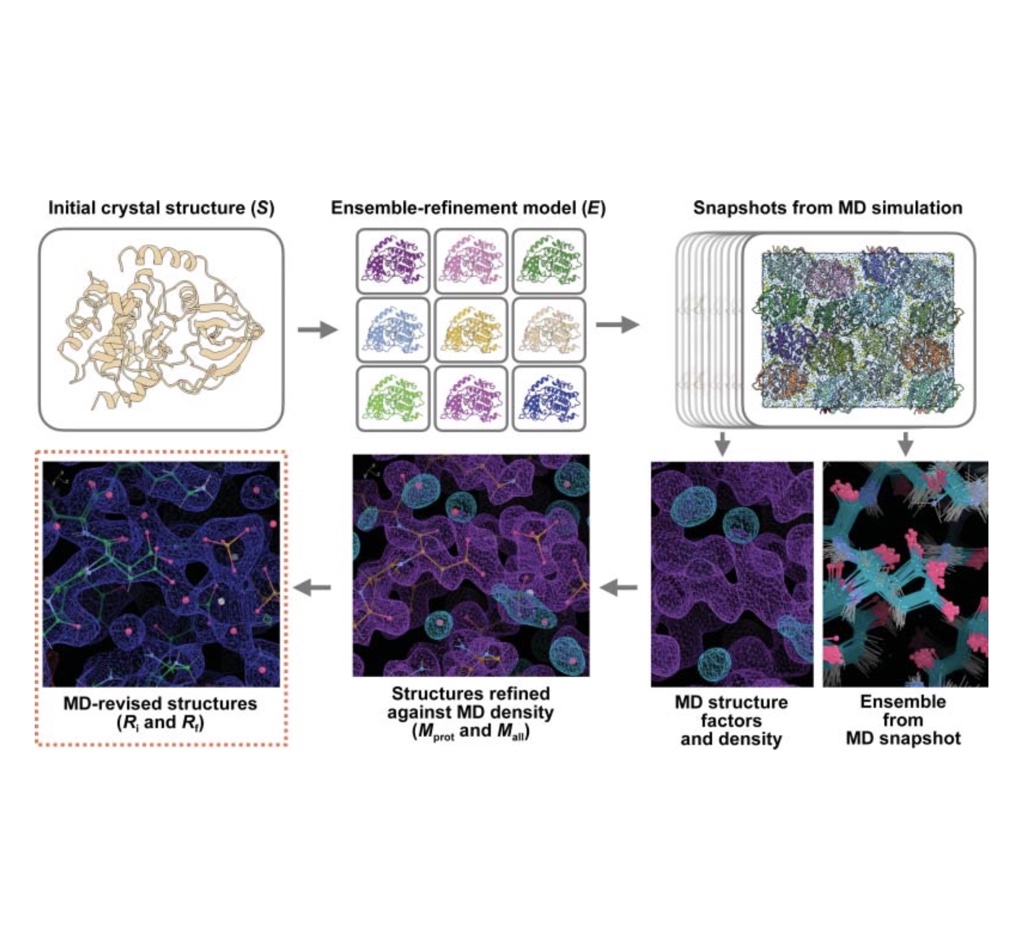
Molecular-dynamics simulation methods for macromolecular crystallography
David C. Wych, Phillip C. Aoto, Lily Vu, Alexander M. Wolff, David L. Mobley, James S. Fraser, Susan S. Taylor, and Michael E. Walla
Preprint ahead of publication: bioRxiv
Published: Structural Biology 79:50, 2023 [DOI]
Code accompanying the publication: jthorton/de-forcefields
We demonstrate how simulations of protein X-ray structures in their crystal environments can be used to assess force field quality
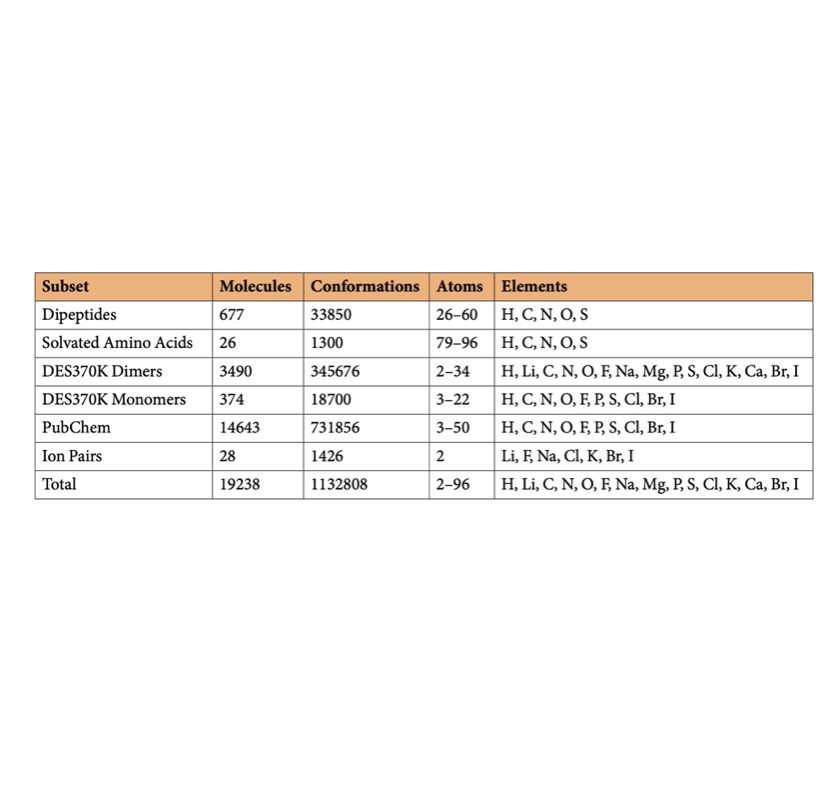
SPICE, A Dataset of Drug-like Molecules and Peptides for Training Machine Learning Potentials
Peter Eastman, Pavan Kumar Behara, David L. Dotson, Raimondas Galvelis, John E. Herr, Josh T. Horton, Yuezhi Mao, John D. Chodera, Benjamin P. Pritchard, Yuanqing Wang, Gianni De Fabritiis, and Thomas E. Markland
Preprint ahead of publication: arXiv
Published: Scientific Data 10:11, 2023 [DOI]
Code accompanying the publication: openmm/spice-dataset
We generate a large open quantum chemical dataset of over 1.1M conformations with broad coverage of biomolecules and druglike small molecules useful for building and assessing force fields.

Development and Benchmarking of Open Force Field 2.0.0: the Sage Small Molecule Force Field
Simon Boothroyd, Pavan Kumar Behara, Owen C. Madin, David F. Hahn, Hyesu Jang, Vytautas Gapsys, Jeffrey R. Wagner, Joshua T. Horton, David L. Dotson, Matthew W. Thompson, Jessica Maat, Trevor Gokey, Lee-Ping Wang, Daniel J. Cole, Michael K. Gilson, John D. Chodera, Christopher I. Bayly, Michael R. Shirts, David L. Mobley
Preprint ahead of publication: chemRxiv
Published: Journal of Chemical Theory and Computation 19:11:3251-3257, 2023 [DOI]
Code accompanying the publication: openforcefield/openff-sage
We introduce Sage (OpenFF 2.0.0), a second-generation small molecule force field for drug-like molecules that includes improved Lennard-Jones parameters.
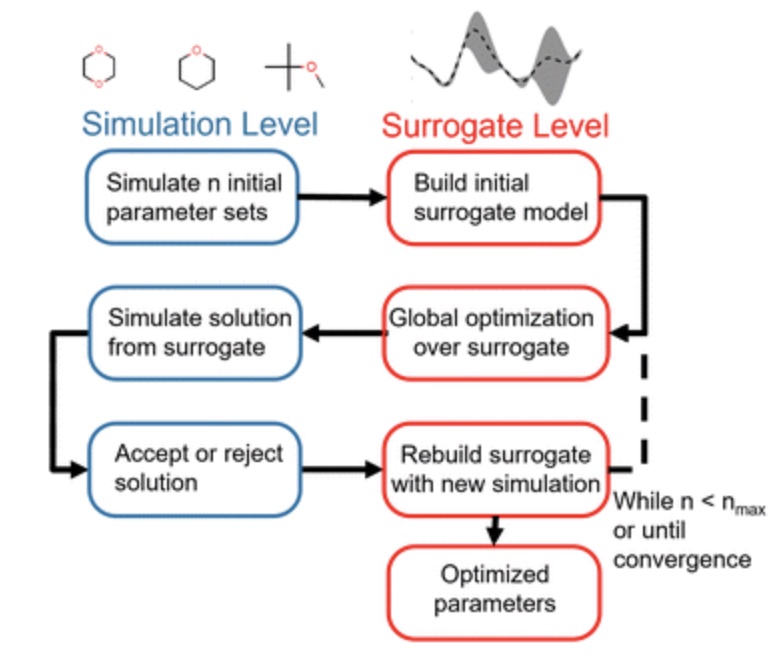
Using physical property surrogate models to perform accelerated multi-fidelity optimization of force field parameters
Owen C. Madin and Michael R. Shirts
Preprint ahead of publication: chemRxiv
Published: Digital Discovery 2:828, 2023 [DOI]
Code accompanying the publication: ocmadin/LJ_surrogates
We demonstrate how fast machine learning surrogate models can be used to accelerate force field parameterization in a multi-fidelity optimization framework
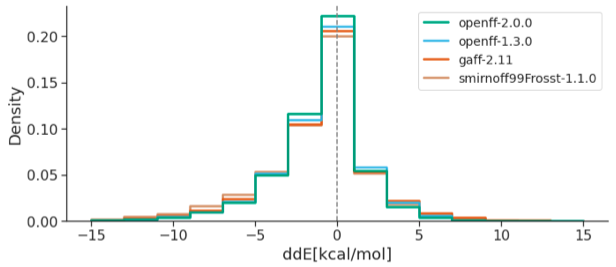
Collaborative assessment of molecular geometries and energies from the Open Force Field
Lorenzo D’Amore, David F. Hahn, David L. Dotson, Joshua T. Horton, Jamshed Anwar, Ian Craig, Thomas Fox, Alberto Gobbi, Sirish Kaushik Lakkaraju, Xavier Lucas, Katharina Meier, David L. Mobley, Arjun Narayanan, Christina E.M. Schindler, William C. Swope, Pieter J. in ’t Veld, Jeffrey Wagner, Bai Xue, and Gary Tresadern
Preprint ahead of publication: chemRxiv CC BY 4.0
Published: Journal of Chemical Information and Modeling 62:23:6094–6104, 2022 [DOI]
Code accompanying the publication: openforcefield/qca-dataset-submission/tree/master/submissions/2021-07-28-OpenFF-Industry-Benchmark-Season-1-MM-v1.1
We present the results of an industry benchmark of OpenFF and other small molecule force fields against quantum chemical data for chemistries of high industrial interest.
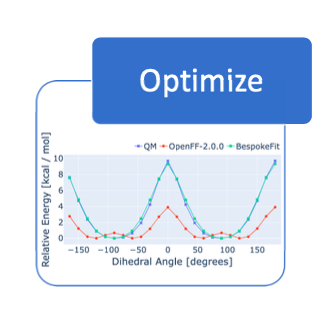
Open Force Field BespokeFit: Automating Bespoke Torsion Parametrization At Scale
Joshua T. Horton, Simon Boothroyd, Jeffery Wagner, Joshua A. Mitchell, Trevor Gokey, David L. Dotson, Pavan Kumar Behara, Venkata Krishnan Ramaswamy, Mark Mackey, John D. Chodera, Jamshed Anwar, David L. Mobley and Daniel J. Cole
Preprint ahead of publication: chemRxiv CC BY 4.0
Published: Journal of Chemical Information and Modeling 62:22:5622–5633, 2022 [DOI]
Code accompanying the publication: openforcefield/openff-bespokefit
We introduce OpenFF BespokeFit, a tool for automating the generation of high-quality bespoke parameters for individual small molecules.
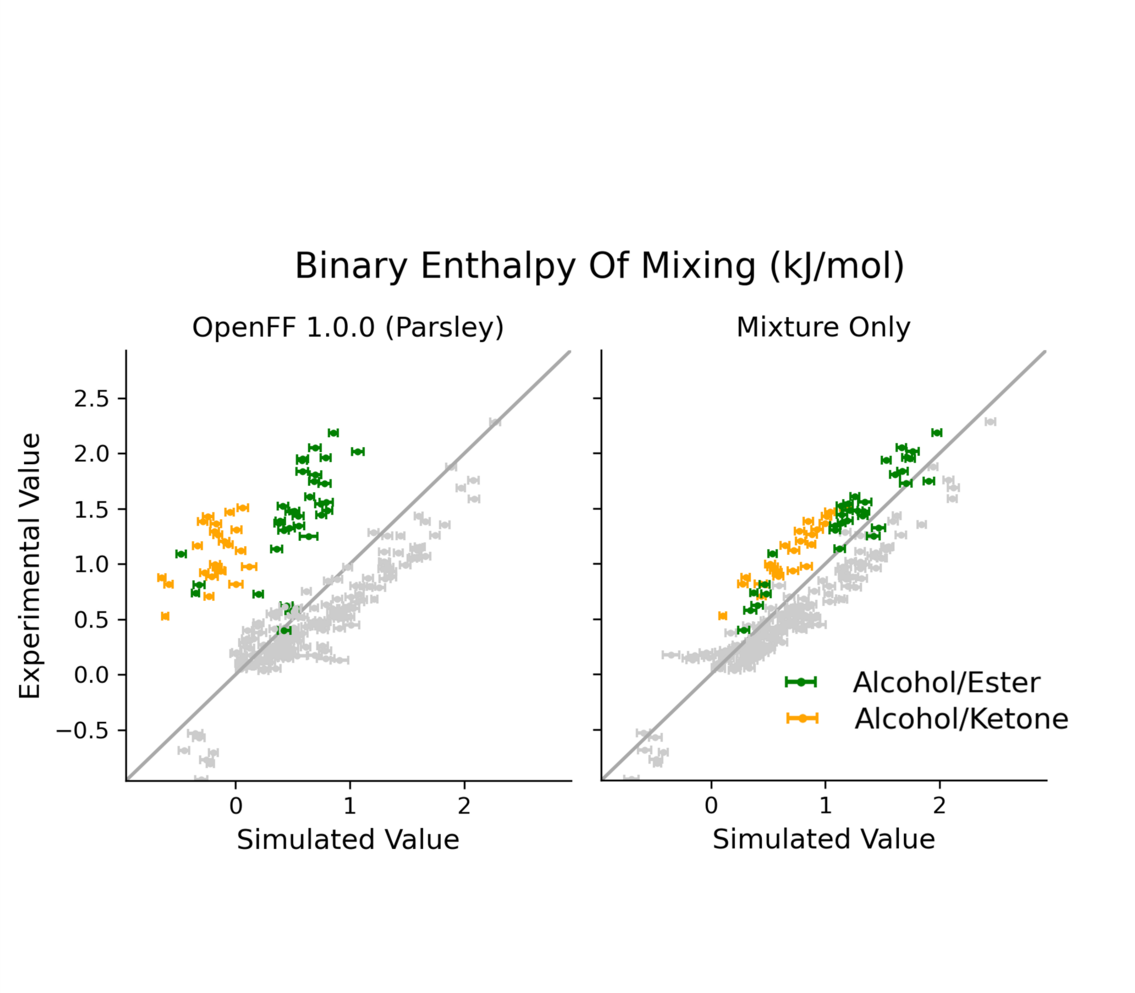
Improving force field accuracy by training against condensed phase mixture properties
Simon Boothroyd, Owen C. Madin, David L. Mobley, Lee-Ping Wang, John D. Chodera and Michael R. Shirts
Preprint ahead of publication: chemRxiv CC BY 4.0
Published: Journal of Chemical Theory and Computation 18:6:3577–3592, 2022 [DOI]
Code accompanying the publication: SimonBoothroyd/binary-mixture-publication
We show how condensed phase mixture properties can be used to substantially improve the quality of biomolecular force fields.
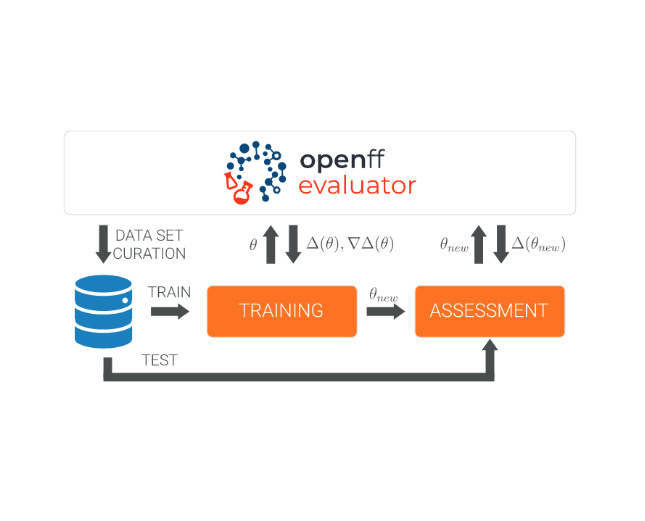
The Open Force Field Evaluator: An automated, efficient, and scalable framework for the estimation of physical properties from molecular simulation
Simon Boothroyd, Lee-Ping Wang, David Mobley, John Chodera, and Michael Shirts
Preprint ahead of publication: chemRxiv CC BY 4.0
Published: Journal of Chemical Theory and Computation 18:6:3566–3576, 2022 [DOI]
Code accompanying the publication: openforcefield/openff-evaluator
We introduce OpenFF Evaluator, an automated tool for estimating physical properties that can be used in both parameterization and assessment pipelines.
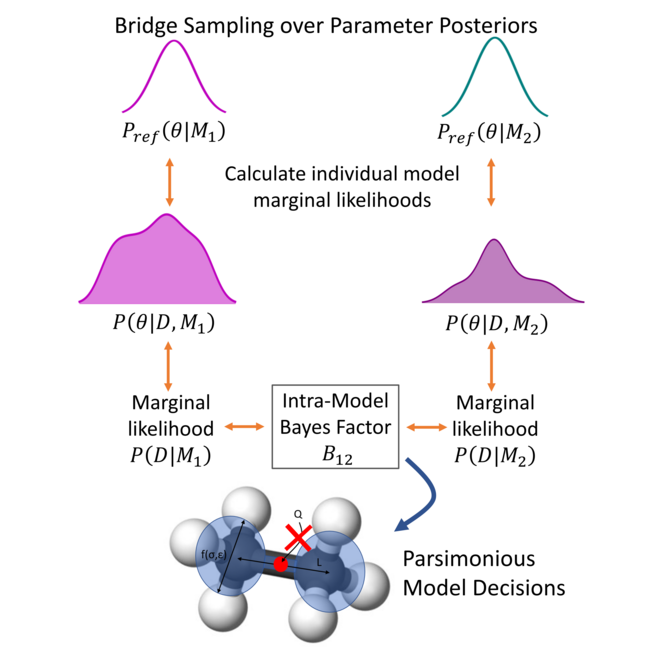
Bayesian inference-driven model parameterization and model selection for 2CLJQ fluid models
Owen C. Madin, Simon Boothroyd, Richard A. Messerly, John D. Chodera, Josh Fass, and Michael R. Shirts
Preprint ahead of publication: arXiv CC BY 4.0
Published: Journal of Chemical Information and Modeling 62:4:874-889, 2022 [DOI]
Code accompanying the publication: SimonBoothroyd/bayesiantesting
We show how Bayesian model selection methods can be used to decide between different functional forms in a statistically rigorous data-driven manner.

Quantum Chemistry Common Driver and Databases (QCDB) and Quantum Chemistry Engine (QCEngine): Automation and interoperability among computational chemistry programs
Daniel G. A. Smith, Annabelle T. Lolinco, Zachary L. Glick, Jiyoung Lee, Asem Alenaizan, Taylor A. Barnes, Carlos H. Borca, Roberto Di Remigio, David L. Dotson, Sebastian Ehlert, Alexander G. Heide, Michael F. Herbst, Jan Hermann, Colton B. Hicks, Joshua T. Horton, Adrian G. Hurtado, Peter Kraus, Holger Kruse, Sebastian J. R. Lee, Jonathon P. Misiewicz, Levi N. Naden, Farhad Ramezanghorbani, Maximilian Scheurer, Jeffrey B. Schriber, Andrew C. Simmonett, Johannes Steinmetzer, Jeffrey R. Wagner, Logan Ward, Matthew Welborn, Doaa Altarawy, Jamshed Anwar, John D. Chodera, Andreas Dreuw, Heather J. Kulik, Fang Liu, Todd J. Martínez, Devin A. Matthews, Henry F. Schaefer, III, Jiří Šponer, Justin M. Turney, Lee-Ping Wang, Nuwan De Silva, Rollin A. King, John F. Stanton, Mark S. Gordon, Theresa L. Windus, C. David Sherrill, and Lori A. Burns
Published: The Journal of Chemical Physics 155:204801, 2021 [DOI]
Code accompanying the publication: https://qcarchive.molssi.org/
We present a community effort to build a common driver and database for quantum chemistry calculations, and demonstrate its use in the QCArchive project.
Best practices for constructing, preparing, and evaluating protein-ligand binding affinity benchmarks [Article v1.0]
David F. Hahn, Christopher I. Bayly, Hannah E. Bruce Macdonald, John D. Chodera, Antonia S. J. S. Mey, David L. Mobley, Laura Perez Benito, Christina E.M. Schindler, Gary Tresadern, Gregory L. Warren
Preprint ahead of publication: arXiv
Published: Living Journal of Computational Molecular Science 4:1:1497, 2022 [DOI]
Code accompanying the publication: openforcefield/FE-Benchmarks-Best-Practices
We present consensus guidelines for (1) curating experimental data to develop meaningful benchmark sets, (2) preparing benchmark inputs according to best practices to facilitate widespread adoption, and (3) analysis of the resulting predictions to enable statistically meaningful comparisons among methods and force fields.
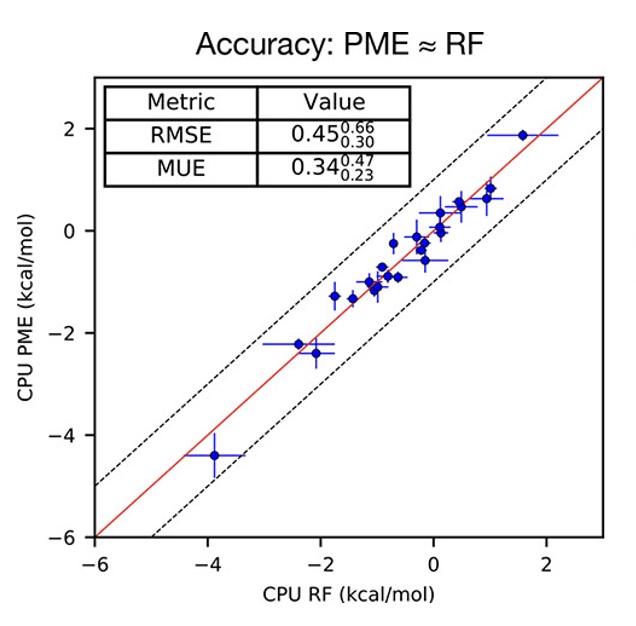
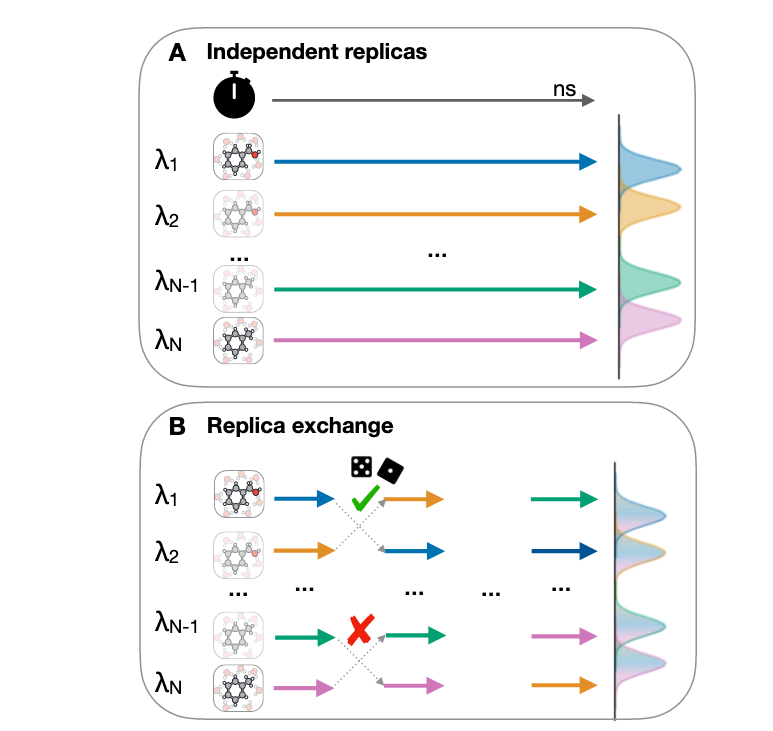
A Benchmark of Electrostatic Method Performance in Relative Binding Free Energy Calculations
Yunhui Ge, David F. Hahn, and David L. Mobley
Preprint ahead of publication: chemRxiv CC BY 4.0
Published: Journal of Chemical Theory and Computation 61:1048, 2021 [DOI]
Code accompanying the publication: openforcefield/openforcefield-forcebalance/tree/v1.0.0-RC2
We compare reaction field (RF) with particle-mesh Ewald (PME) models of electrostatics for protein-ligand complex relative binding free energies.

Development and Benchmarking of Open Force Field v1.0.0, the Parsley Small Molecule Force Field
Yudong Qiu, Daniel Smith, Simon Boothroyd, Hyesu Jang, Jeffrey Wagner, Caitlin C. Bannan, Trevor Gokey, Victoria T. Lim, Chaya Stern, Andrea Rizzi, Xavier Lucas, Bryon Tjanaka, Michael R. Shirts, Michael Gilson, John Chodera, Christopher I. Bayly, David Mobley, Lee-Ping Wang
Preprint ahead of publication: chemRxiv CC BY 4.0
Published: Journal of Chemical Theory and Computation 17:10:6262-6280, 2021 [DOI]
Code accompanying the publication: openforcefield/openforcefield-forcebalance/tree/v1.0.0-RC2
We present the structure and optimization of the first-generation Parsley (OpenFF 1.0.0) small molecule force field, which uses the SMIRKS-native Open Force Field (SMIRNOFF) parameter assignment formalism to achieve high accuracy with minimal parameters.
End-to-end differentiable construction of molecular mechanics force field
Yuanqing Wang, Josh Fass, Benjamin Kaminow, John E. Herr, Dominic Rufa, Ivy Zhang, Iván Pulido, Mike Henry, Hannah E. Bruce Macdonald, Kenichiro Takaba and John D. Chodera
Preprint ahead of publication: arXiv CC BY 4.0
Published: Chemical Science 13:12016-12033, 2022 [DOI]
Code accompanying the publication: choderalab/espaloma
Molecular mechanics force fields have been a workhorse for computational chemistry and drug discovery. Here, we propose a new approach to force field parameterization in which graph convolutional networks are used to perceive chemical environments and assign molecular mechanics (MM) force field parameters. The entire process of chemical perception and parameter assignment is differentiable end-to-end with respect to model parameters, allowing new force fields to be easily constructed from MM or QM force fields, extended, and applied to arbitrary biomolecules.
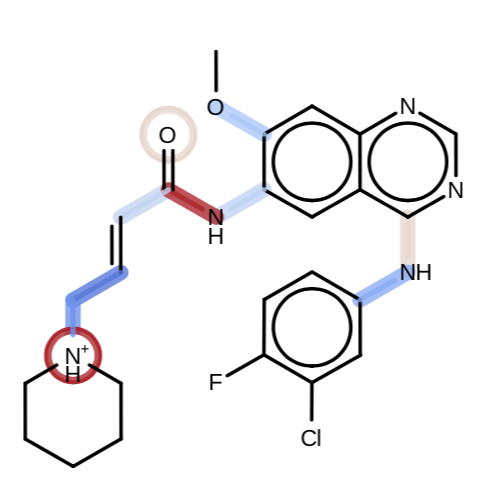
Capturing non-local through-bond effects when fragmenting molecules for quantum chemical torsion scans
Chaya D. Stern, Christopher I. Bayly, Daniel G. A. Smith, Josh Fass, Lee-Ping Wang, David L. Mobley, and John D. Chodera
Preprint ahead of publication: bioRxiv CC BY 4.0
Code accompanying the publication: openforcefield/fragmenter
We show how the Wiberg Bond Order (WBO) can be used to construct small molecule fragmentation schemes that will avoid disrupting the chemical environment around torsions. The resulting fragmentation scheme powers the QCSubmit tool used to fragment and inject small molecule datasets into the QCFractal computation pipeline for deposition into the QCArchive quantum chemistry archive the Open Force Field Initiative uses for constructing force fields, as well as powering bespoke torsion refitting for individual molecules.
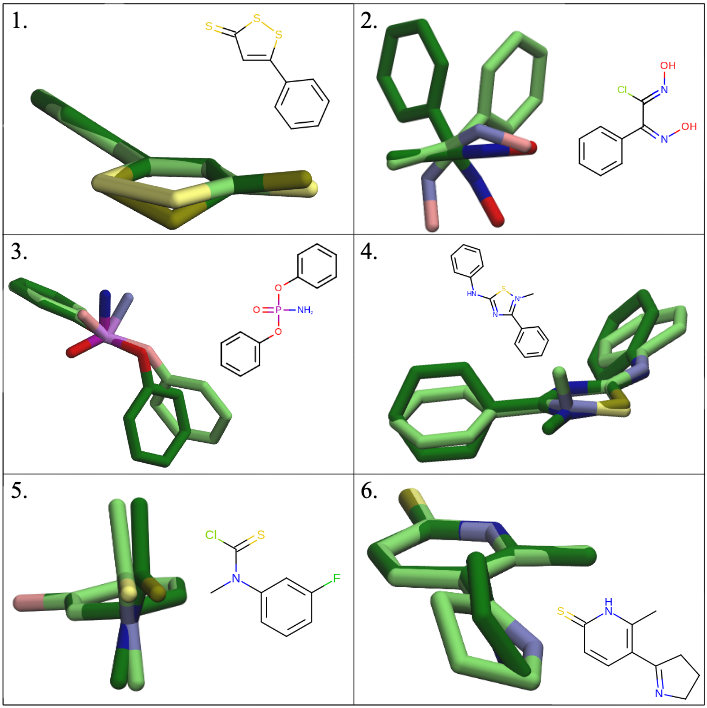
Improving Small Molecule Force Fields by Identifying and Characterizing Small Molecules with Inconsistent Parameters
Jordan Ehrman, Victoria T. Lim, Caitlin C. Bannan, Nam Thi, Daisy Kyu, and David Mobley
Preprint ahead of publication: chemRxiv CC BY 4.0
Published: Journal of Computer-Aided Molecular Design 35:271-284, 2021 [DOI]
Code accompanying the publication: mobleylab/off-ffcompare
We present a pipeline for comparing the geometries of small molecule conformers optimized with different force fields. We aimed to identify molecules or chemistries that are particularly informative for future force field development because they display inconsistencies between force fields. We applied our pipeline to a subset of the eMolecules database, and highlighted molecules that appear to be parameterized inconsistently across different force fields. We then identified over-represented functional groups in these molecule sets. The molecules and moieties identified by this pipeline may be particularly helpful for future force field parameterization.
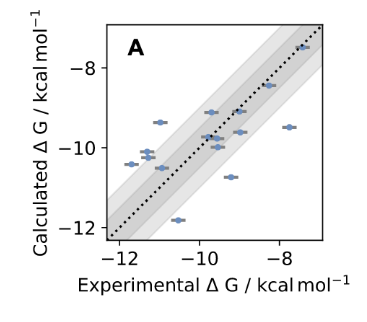
Towards chemical accuracy for alchemical free energy calculations with hybrid physics-based machine learning/molecular mechanics potentials
Dominic Rufa, Hannah Bruce Macdonald, Josh Fass, Marcus Wieder, Patrick Grinaway, Adrian Roitberg, Olexandr Isayev and John Chodera
Preprint ahead of publication: bioRxiv CC BY 4.0
Code accompanying the publication: choderalab/perses
This study combines a new generation of hybrid ML/MM potentials and a nonequilibrium perturbation approach to predict protein-ligand binding affinities. With this approach, a standard, GPU-accelerated MM alchemical free energy calculation can be corrected in a simple post-processing step to efficiently recover ML/MM free energies, while delivering a significant accuracy improvement with small additional computational effort. The authors show that it is possible to significantly reduce the error in absolute binding free energies with this new hybrid ML/MM approach ANI2xx/AMBER14SB/TIP3P on Tyk2 benchmarking system. The same set of FE calculations performed with OpenFF-1.0.0 instead of ANI2xx to model ligands achieves RMSE statistically indistinguishable from the Schrodinger JACS result for the tested system, which implies that we should expect even better results with the latest Parsley update (OpenFF-1.2.0).
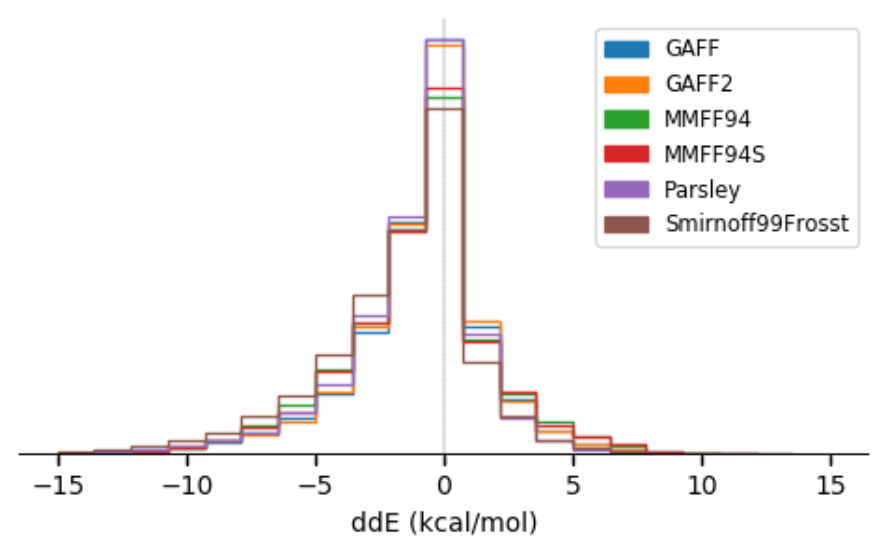
Benchmark Assessment of Molecular Geometries and Energies from Small Molecule Force Fields
Victoria T. Lim and David L. Mobley
Preprint ahead of publication: chemRxiv CC BY 4.0
Published: F1000Research 9:1390, 2020 [DOI]
Code accompanying the publication: mobleylab/benchmarkff
In this work, we aim to compare six force fields: GAFF, GAFF2, MMFF94, MMFF94S, SMIRNOFF99Frosst, and the openff-1.0.0 (Parsley) force field by focusing on small molecules (< 50 heavy atoms). On a dataset comprising over 26,000 molecular structures, we analyzed their force field-optimized geometries and conformer energies compared to reference quantum mechanical (QM) data. We show that most of these force fields are comparable in accuracy at reproducing gas-phase QM geometries and energetics, but that GAFF/GAFF2/Parsley do slightly better in reproducing QM energies and that MMFF94/MMFF94S perform slightly better in geometries. Parsley version OpenFF-1.0.0 shows considerable improvement over its predecessor SMIRNOFF99Frosst, while OpenFF-1.2.0 performs even better with accuracy comparable to other available general force fields. We identify particular outlying chemical groups for further force field improvement.
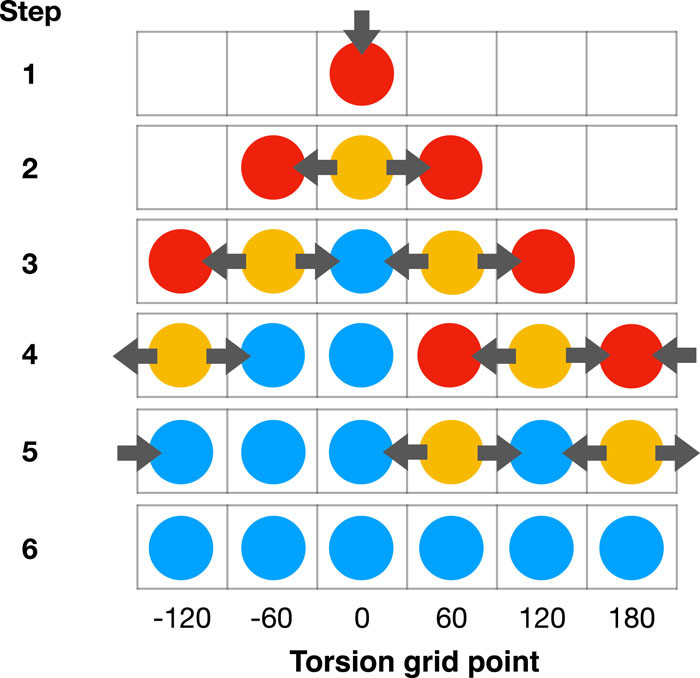
Driving torsion scans with wavefront propagation
Yudong Qiu, Daniel G. A. Smith, Chaya D. Stern, Mudong Feng, Hyesu Jang, and Lee-Ping Wang
Preprint ahead of publication: chemRxiv CC BY 4.0
Published: The Journal of Chemical Physics 152:244116, 2020 [DOI]
Code accompanying the publication: lpwgroup/torsiondrive/
In this paper, we propose a systematic and versatile workflow called TorsionDrive to generate energy-minimized structures on a grid of torsion constraints by means of a recursive wavefront propagation algorithm, which resolves the deficiencies of conventional scanning approaches and generates higher quality QM data for force field development. The method is implemented in an open-source software package that is compatible with many QM software packages and energy minimization codes. The paper also describes integration with the MolSSI QCArchive distributed computing ecosystem.

Binding thermodynamics of host-guest systems with SMIRNOFF99Frosst 1.0.5 from the Open Force Field Initiative
David R. Slochower, Niel Henriksen, Lee-Ping Wang, John D. Chodera, David L. Mobley, and Michael K. Gilson
Preprint ahead of publication: chemRxiv CC BY 4.0
Published: Journal of Chemical Theory and Computation 15:6225, 2019 [DOI]
Code accompanying the publication: slochower/smirnoff-host-guest-manuscript
We evaluate the accuracy of SMIRNOFF99Frosst, using free energy calculations of 43 α and β-cyclodextrin host-guest pairs and compare with matched calculations using two versions of GAFF. These results suggest that SMIRNOFF99Frosst performs competitively with existing small molecule force fields and is a parsimonious starting point for optimization.
Graph Nets for Partial Charge Prediction
Yuanqing Wang and Josh Fass and Chaya D. Stern and John Chodera
Preprint ahead of publication: arXiv arXiv-1.0
Code accompanying the publication: choderalab/gimlet
Graph convolutional and message-passing networks can be a powerful tool for predicting physical properties of small molecules when coupled to a simple physical model that encodes the relevant invariances. Here, we show the ability of graph nets to predict partial atomic charges for use in molecular dynamics simulations and physical docking.
ChemPer: An Open Source Tool for Automatically Generating SMIRKS Patterns
Caitlin C. Bannan, David Mobley
Preprint ahead of publication: chemRxiv CC BY 4.0
Code accompanying the publication: MobleyLab/chemper
In this work, we present ChemPer – a new tool for generating SMIRKS patterns based on clustered fragments (i.e. bonds, angles, or torsions) which should be assigned the same force field parameter. We demonstrate the utility of ChemPer by clustering fragments based on a reference force field, and then regenerating those parameters starting with a simple set of alkanes, ethers, and alcohols.
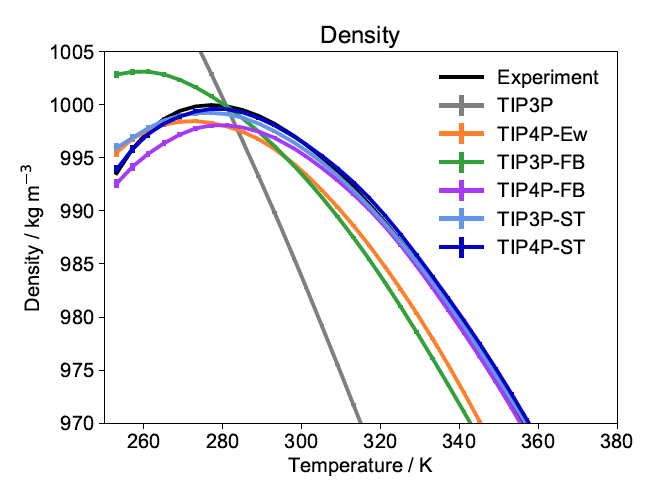
Systematic Optimization of Water Models Using Liquid/Vapor Surface Tension Data
Yudong Qiu, Paul S. Nerenberg, Teresa Head-Gordon, Lee-Ping Wang
Preprint ahead of publication: chemRxiv CC BY 4.0
Published: The Journal of Physical Chemistry B 123:7061, 2019 [DOI]
Code accompanying the publication: leeping/forcebalance
This work investigates whether experimental surface tension measurements, which are less sensitive to quantum and self-polarization corrections, are able to replace the usual reliance on the heat of vaporization as experimental reference data for fitting force field models of molecular liquids.
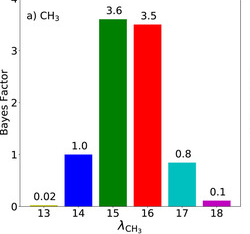
Uncertainty quantification confirms unreliable extrapolation toward high pressures for united-atom Mie λ-6 force field
Richard A. Messerly, Michael R. Shirts, and Andrei F. Kazakov
Published: The Journal of Chemical Physics 149:114109, 2018 [DOI]
We demonstrate how Bayesian approaches can be used to estimate the reliability of predictions made with molecular mechanics force fields.
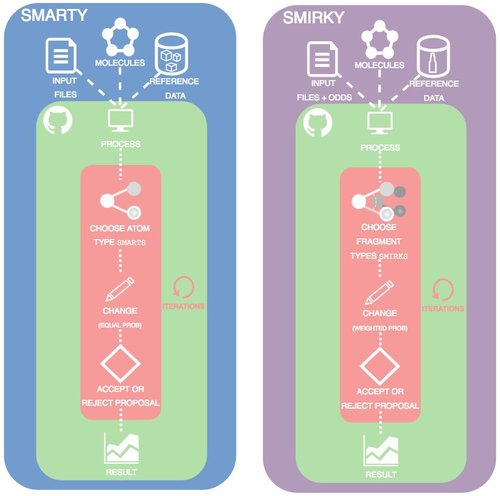
Toward learned chemical perception of force field typing rules
Camila Zanette, Caitlin C. Bannan, Christopher I. Bayly, Josh Fass, Michael K. Gilson, Michael R. Shirts, John Chodera, and David L. Mobley
Preprint ahead of publication: chemRxiv CC BY 4.0
Published: Journal of Chemical Theory and Computation 15:402, 2019 [DOI]
Code accompanying the publication: openforcefield/smarty
Here, we introduce a proof of principle for automatically sampling chemical perception compared to traditional atom typed force fields and our SMIRNOFF format.
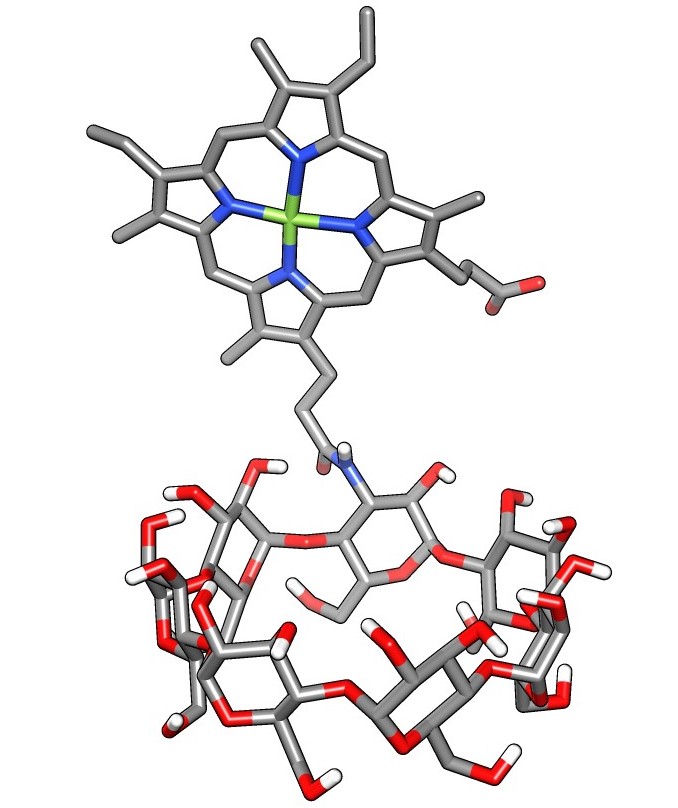
Facile Synthesis of a Diverse Library of Mono-3-substituted β-Cyclodextrin Analogues
Kathryn Kellett, Brendan M. Duggan and Michael K. Gilson
Preprint ahead of publication: chemRxiv CC BY 4.0
Published: Supramolecular Chemistry 31:251, 2019 [DOI]
We show the facile synthesis of a library of diverse mono-3-substituted β-cyclodextrin analogues, that have the potential to be used to collect host-guest binding data to test and improve simulation force fields.
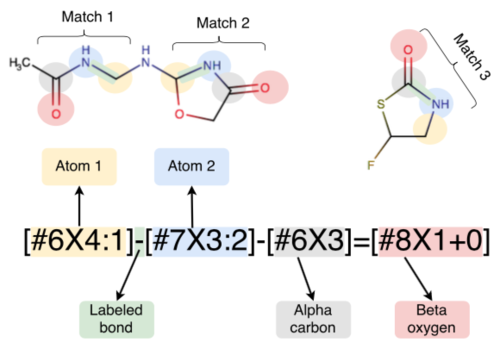
Escaping atom types using direct chemical perception
David Mobley, Caitlin C. Bannan, Andrea Rizzi, Christopher I. Bayly, John D. Chodera, Victoria T Lim, Nathan M. Lim, Kyle A. Beauchamp, Michael R. Shirts, Michael K. Gilson, and Peter K. Eastman
Preprint ahead of publication: bioRxiv CC BY 4.0
Published: Journal of Chemical Theory and Computation 14:6076, 2018 [DOI] PMC6245550
This paper introduces the SMIRNOFF format in the context of traditional force fields, explains the development and validation of our new small molecule force field smirnoff99Frosst, and highlights some directions the initiative is headed.
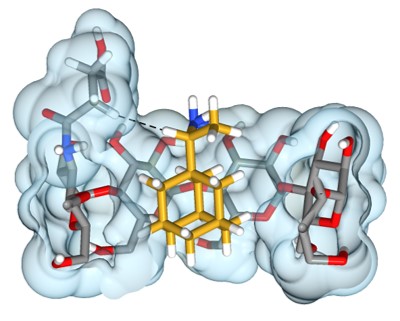
Toward Expanded Diversity of Host–Guest Interactions via Synthesis and Characterization of Cyclodextrin Derivatives
Kathryn Kellett, S. A. Kantonen, Brendan M. Duggan and Michael K. Gilson
Preprint ahead of publication: chemRxiv CC BY-NC-ND 4.0
Published: The Journal of Solution Chemistry 47:1597, 2018 [DOI]
This paper shows the synthesis of a mono-3-functionalized cyclodextrin and how cyclodextrin derivatives can effect the binding of guest molecules using 1D/2D NMR and ITC experiments.
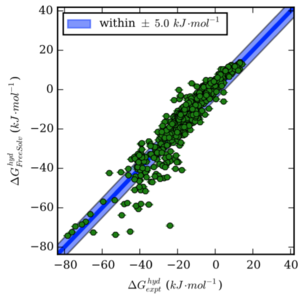
Approaches for Calculating Solvation Free Energies and Enthalpies Demonstrated with an Update of the FreeSolv Database
Guilherme Duarte Ramos Matos, Daisy Y. Kyu, Hannes H. Loeffler, John D. Chodera, Michael R. Shirts, and David L. Mobley
Preprint ahead of publication: bioRxiv CC BY 4.0
Published: Journal of Chemical Engineering Data 62:1559, 2017 [DOI] PMC5648357
Code accompanying the publication: mobleylab/freesolv
We review alchemical methods for computing solvation free energies and present an update (version 0.5) to the FreeSolv database of experimental and calculated hydration free energies of neutral compounds.
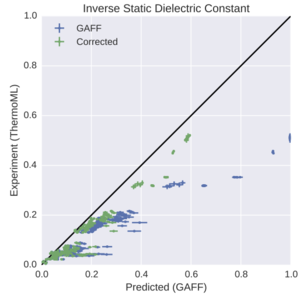
Towards Automated Benchmarking of Atomistic Forcefields: Neat Liquid Densities and Static Dielectric Constants from the ThermoML Data Archive
Kyle A. Beauchamp, Julie M. Behr, Ariën S. Rustenburg, Christopher I. Bayly, Kenneth Kroenlein, and John D. Chodera
Preprint ahead of publication: arXiv arXiv-1.0
Published: Journal of Physical Chemistry B 119:12912, 2015 [DOI] PMC4667959
Code accompanying the publication: choderalab/LiquidBenchmark
Progress in forcefield validation and parameterization has been hindered by the availability of high-quality machine-readable physical property data for small organic molecules. We show how the NIST ThermoML dataset provides a solution to this problem, and demonstrate its utility in benchmarking the GAFF/AM1-BCC small molecule forcefield on neat liquid densities and static dielectric constants to uncover problems in the representation of low-dielectric environments.