Self-consistently simulating small molecules, proteins and RNAs with the "espaloma-0.3" force field
Posted on 12 Feb 2024 by Yuanqing Wang

The Espaloma force fields are a class of graph neural networks (GNNs)-parametrized molecular mechanics (MM) force fields where the human-driven, labor-intensive, inflexible, and inextensible atom types are replaced by machine-learned representations, which are subsequently composed to yield the per-atom, -bond, -angle, and -torsion parameters. When training, we can take derivatives of the energies and forces with regard to all relative force field parameters and neural network parameters, optimizing them against quantum chemical data to approach quantum chemical accuracy; during inference, since the functional forms remain the same, they are exactly as fast as a traditional MM force field.
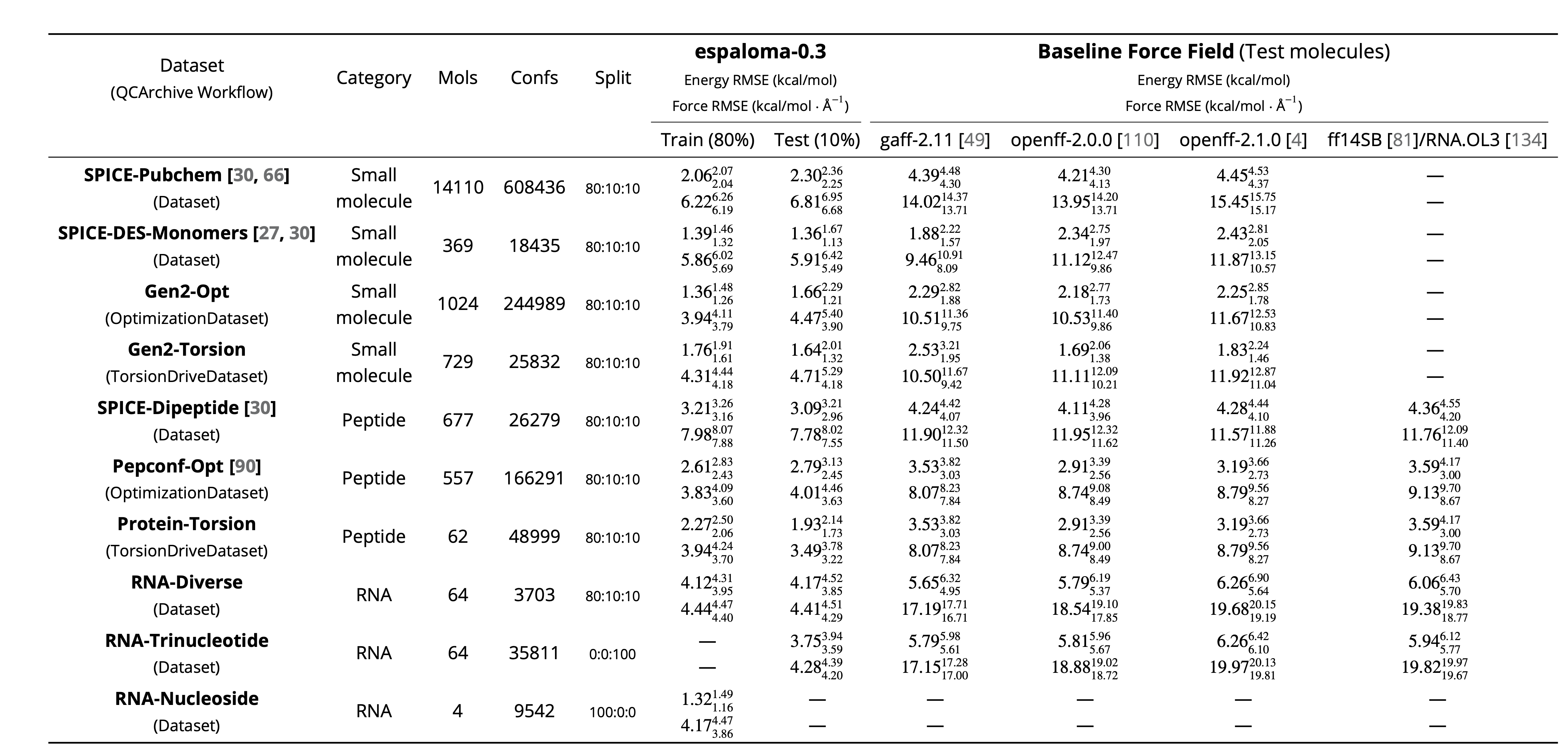
Up until recently, Espaloma remained a prototype trained only on quantum chemical calculations of little over one thousand molecules. Now, there is a new generation of Espaloma, christened espaloma-0.3, covering vast chemical spaces of potential interest to medicinal chemists, from small molecules and proteins to RNAs. Moreover, force-matching (where we tune the force field to reproduce the quantum chemical forces) has also been incorporated into this version of espaloma, making the force fields more accurate and stable.
Here is a table summarizing the chemical space coverage of espaloma-0.3, together with the energy and force accuracy:
Installation
espaloma-0.3, just like many other essential tools in computational chemistry, is on conda-forge! It can be easily installed by:
$ conda install -c conda-forge "espaloma=0.3.2"
Deployment
To deploy espaloma-0.3 is similarly easy—you would just have to massage your topology into one from openff.toolkit and espaloma can take care of the rest to return an openmm.System object:
# import
import espaloma as esp
# define or load a molecule of interest via the Open Force Field toolkit
from openff.toolkit.topology import Molecule
molecule = Molecule.from_smiles("CN1C=NC2=C1C(=O)N(C(=O)N2C)C")
# create an Espaloma Graph object to represent the molecule of interest
molecule_graph = esp.Graph(molecule)
# load pretrained model
espaloma_model = esp.get_model("latest")
# apply a trained espaloma model to assign parameters
espaloma_model(molecule_graph.heterograph)
# create an OpenMM System for the specified molecule
openmm_system = esp.graphs.deploy.openmm_system_from_graph(molecule_graph)
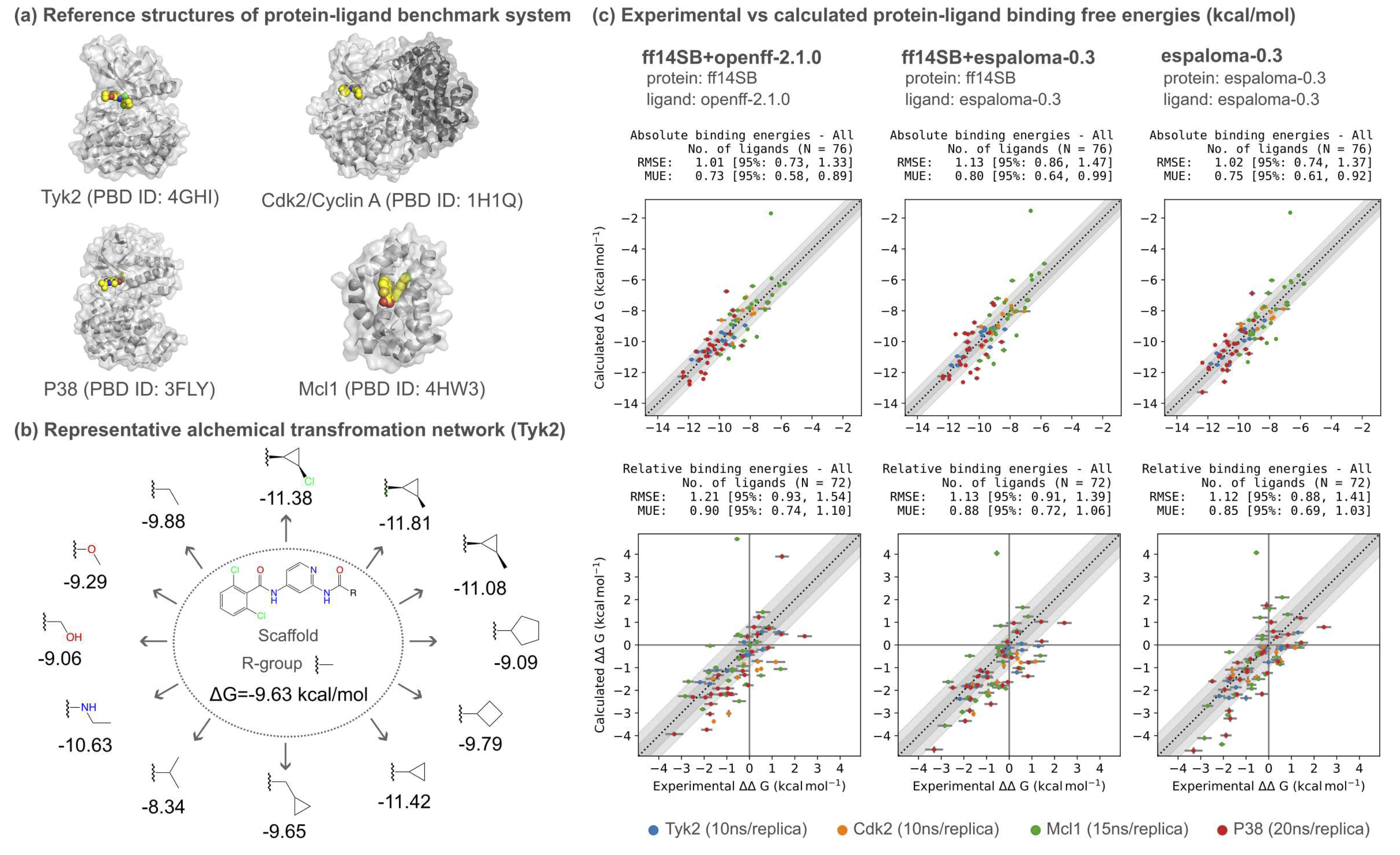
Applications: protein-ligand binding free energy calculations
We have tested a few applications you can try out using the new espaloma-0.3 force field, for example, protein-ligand binding free energy prediction.