
Webinars
Watch all currently available webinars on our YouTube channel. You can access slides on Zenodo. Please use the assigned DOI and associated publications if you plan on using provided materials in your work.
If you are interested in presenting your work in our OFF webinar series, please fill out this form. We will share your talk on our YouTube channel and on our social media networks. We accept various topics relevant to force field development and molecular dynamics simulations.
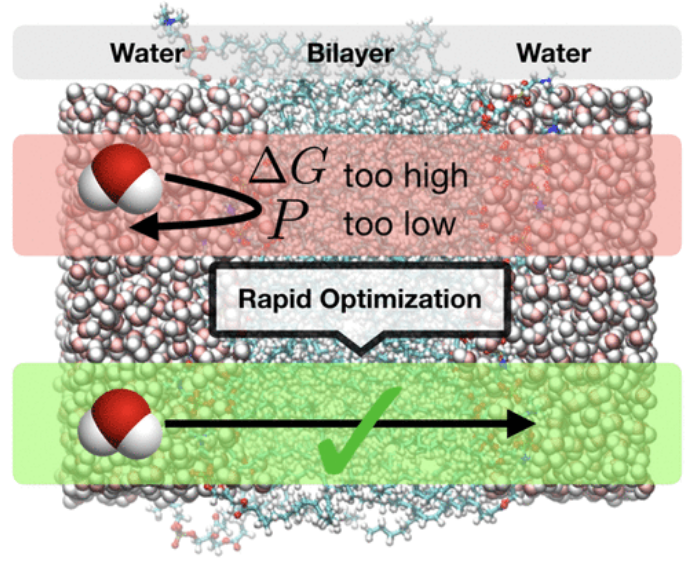
Automated Optimization Approaches for the CHARMM Lipid Force Field
Andreas Krämer (NIH) gave a talk about development of an automated optimization approach to improve the CHARMM36 lipid force field using pair-specific LJ parameters during his visit to MSKCC.
The CHARMM36 (C36) lipid force field is well-validated for many properties of monolayers and bilayers and has been cited in over 2000 studies of membrane simulations. Problematic aspects of C36 include its hard-wired fine-tuning to the TIP3P water model as well as intricacies regarding the Lennard-Jones (LJ) interactions. Concretely, the LJ switch distance is inconsistent between the C36 protein and lipid force fields and monolayer surface tensions only agree with experiment when long-range dispersion is incorporated. In contrast, bilayer properties were optimized without long-range dispersion.
To improve this situation, we first explore the water-model dependence and develop an automated optimization approach to better reproduce membrane permeabilities using pair-specific LJ parameters. We then carry out a reweighting-based optimization of the force field to consistently introduce long-range dispersion. The initial results for 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) show that within few iterations the match with experiment is improved for many important lipid properties, such as bilayer and monolayer surface areas, compressibilities, order parameters, and headgroup hydration.

Graph Nets for partial charge prediction
Yuanqing Wang (MSKCC) gave a talk about using Graph Nets for fast prediction of atomic partial charges. The preprint is available here.
Here we show that Graph Nets — a set of update and aggregate functions that operate on molecular topologies and propagate information thereon — are capable of predicting properties of atoms and molecules which would otherwise require expensive Quantum Mechanics (QM) calculations. This architecture is applied to predict atomic partial charges, which are crucial parameters for Molecular Dynamics (MD) simulations, molecular mechanics calculations, and virtual screening, as they determine the electrostatic contributions to interaction energies. We will also briefly introduce our work-in-progress on using this model for inter-hierarchical multitask learning and on Hypergraph Functional Potential (HGFP) — a fast and flexible machine learning force field.
Using NMR relaxation data to improve the dynamics of methyl groups in AMBER and CHARMM force fields
Falk Hoffman from Ruhr University Bochum presented his work on improving torsional barriers associated with methyl group rotations in amino acid side-chains in AMBER and CHARMM force fields by using NMR relaxation rates. This study is available as a preprint on ChemrXiv.
The internal dynamics of proteins occurring on time scales from picoseconds to nanoseconds can be sensitively probed by nuclear magnetic resonance (NMR) spin relaxation experiments, as well as by molecular dynamics (MD) simulations. This complementarity offers unique opportunities, provided that the two methods are compared at a suitable level. Recently, several groups have used MD simulations to compute the spectral density of backbone and side-chain molecular motions, and to predict NMR relaxation rates from these. Unfortunately, in the case of methyl groups in protein side-chains, inaccurate energy barriers to methyl rotation were responsible for a systematic discrepancy in the computed relaxation rates.
In this talk I will present how we used NMR relaxation rates to identify disagreements between simulation and experiment for these energy barriers. The corresponding dihedral angle terms in AMBER and CHARMM force fields which are responsible for these energy barriers are subsequently improved and correspondence between experiment and simulation could be regained by amending the MD force field with accurate coupled cluster quantum chemical calculations. Improved methyl group rotation barriers were derived, such that the NMR relaxation data obtained from the MD simulations now also display very good agreement with experiment. Furthermore, I will show how these energy barriers influence the validity of the Lipari-Szabo model for protein side-chains which is used to extract generalized order parameters from NMR relaxation experiments and MD simulations.

Data curation - the forgotten practice in the era of AI
Pankaj R. Daga from Simulation-Plus visited the Mobley group at UC Irvine and gave a talk about all the hazards that can emerge in attempts to automate mining of chemical and chemistry-related databases.
Availability of large databases of chemical structures along with experimental data provides a great opportunity to build predictive and robust QSAR/QSPR models for application in various fields. The most common concern while using these databases is the quality of the chemical structures and associated biological data. It is very important to deal with correct chemical structure since incorrect structure will lead to the errors in calculation of molecular descriptors. Incorrect biological data will ultimately lead to meaningless results. This seminar will discuss experiences while curating these bioactivity databases with focus towards ADMET properties in drug discovery. Various sources of these errors and measures to find and correct these errors will be discussed.
ff19SB: Amino acid specific protein backbone parameters trained against quantum mechanics energies in solution
Chuan Tian from the Simmerling lab visited the Chodera lab at MSKCC on Sep 12, 2019 and gave a talk about the latest Amber force field - ff19SB.
Molecular dynamics (MD) simulations have become increasingly popular in studying the motions and functions of biomolecules. The accuracy of the simulation, however, is highly determined by the classical force field (FF), a set of functions with adjustable parameters to compute the potential energy from atomic positions. The relatively simple terms in most of the current force fields are computationally advantageous which enable the simulation of biologically important macromolecules at biologically relevant timescale. However, the overall quality of the FF, including our previously published ff14SB, is limited by the assumptions that were made years ago. (1) An overly symmetric φ/ψ dihedral energy map arises from the uncoupled cosine functions used to model these two degrees of freedom in the protein backbone. (2) The model does not show sufficient dependence of the backbone energetics on the amino acids, probably because the parameters developed for the simple amino acid Ala were applied to all other amino acids without checking the quality of the transferability. (3) The fixed partial charges were trained for aqueous solution, but the dihedral parameters were all fit to gas-phase QM, thus the resulting dihedral parameters actually counteract the intended polarization effect and introduce significant internal inconsistency in the model.
In our new force field ff19SB, we have significantly improved the backbone profiles for all 20 amino acids. We have developed an amino-acid specific CMAP term by fitting against in-solution QM data. Our results show that the new FF not only better reproduces various types of experimental data on amino-acid specific properties such as NMR scalar coupling and helical propensity, but it also improves secondary structure content in small peptides and maintains reasonably accurate protein NMR order parameters.
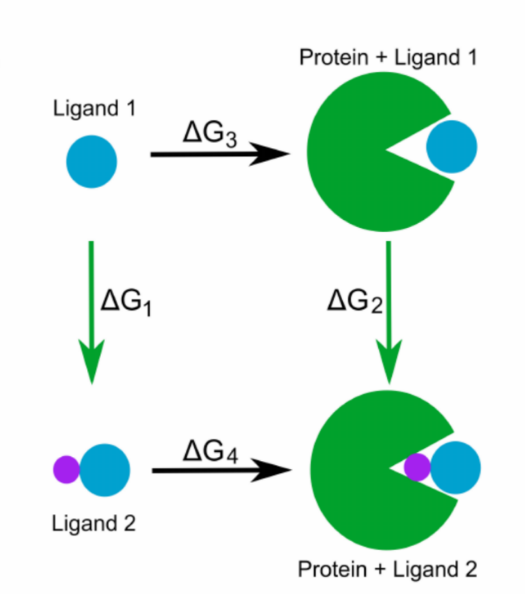
Binding free energy calculations for protein-ligand systems
Vytautas Gapsys was hosted by the Mobley lab at UC Irvine on Aug 21, 2019, where he presented his work on calculations of relative ligand-protein affinities using Gromacs+pmx approach. His talk also covered a free energy scan based on the non-equilibrium protocol covering ~500 ligand modifications across 13 protein-ligand systems, and provided comparison of the obtained results to FEP+ calculations.
Fragmenting molecules for QC torsions
Chaya Stern talks about molecular fragmentation prior to computing QM torsion profiles and using Wiberg bond orders as potentially powerful indicators of change in chemical environment of rotatable bonds upon fragmentation.